
ASTM E2500-25:以科学风险为基础的制药设备验证新范式——覆盖全生命周期的合规演进
本文包含AI辅助创作内容
在制药与生物制药行业,生产系统与设备的合规性是保障产品质量和患者安全的核心基石。ASTM 国际组织发布的《ASTM E2500-25 制药和生物制药生产系统与设备的规范、设计和验证标准指南》,以 “科学为基、风险为核” 的理念,构建了覆盖设备全生命周期的合规框架,为企业从设计到退役的全流程操作提供了明确指引。本文将深度拆解标准核心要求,结合原文规范、实操要点及关键图表,帮助大家实现从 “合规达标” 到 “质量可控” 的进阶。
ASTM E2500-25 并非局限于单一设备或环节的验证规范,其核心价值在于建立了一套系统化、全生命周期的管理体系。
标准原文明确界定适用边界:
"This guide is applicable to all elements of pharmaceutical and biopharmaceutical manufacturing systems including: good manufacturing practice (GMP) utility equipment, process equipment, supporting utilities, associated process monitoring and control systems,and automation systems that have the potential to affect product quality, availability, and/or patient safety."
指南适用于制药和生物制药生产系统的所有要素,包括药品生产质量管理规范(GMP)公用设施设备、工艺设备、辅助公用设施、相关工艺监测和控制系统,以及可能影响产品质量、可用性和 / 或患者安全的自动化系统。
从覆盖范围来看,标准不仅适用于新建系统,也包含现有系统的变更与维护,甚至可延伸至实验室设备、医疗器械生产系统(实验室仪器确认参考 USP <1058>),真正实现 “全场景覆盖”;从生命周期来看,其要求贯穿系统 “概念设计 - 规格制定 - 验证确认 - 运行维护 - 退役处置” 全阶段,形成闭环管理;从核心目标来看,所有要求均围绕 “产品关键质量属性(CQA)” 与 “患者安全” 展开,风险管控贯穿始终。
标准开篇明确了 9 个核心术语定义,是理解合规要求的前提,其中最易混淆的 3 组概念如下: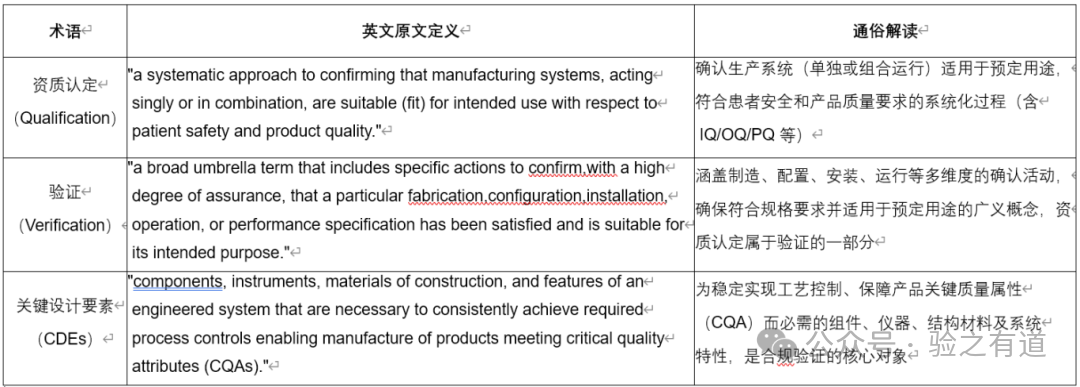
此外,标准还明确了关键工艺参数(CPP)、质量风险管理(QRM)、系统所有者(System Owner)等核心术语,为后续操作提供统一的定义基准。例如,“关键工艺参数(CPP)” 被定义为 “其变异性会影响关键质量属性,因此需监测或控制以确保工艺产出预期质量” 的参数,这一概念直接关联后续验证活动的范围划定。
核心框架:6 大关键概念
ASTM E2500-25 的核心逻辑围绕 6 大关键概念展开,形成 “要求 - 设计 - 验证 - 改进” 的闭环,而标准中的核心图表(则将抽象逻辑转化为可视化流程,是理解合规路径的关键。
"The evaluation of the risk to quality should be based on scientific knowledge and ultimately link to the protection of the patient."(质量风险评估应基于科学知识,并最终与患者保护相关联。)
风险评估的结果直接决定验证的范围和深度 —— 高风险环节(如直接接触药品的生产设备)需更严格的验证流程和文档要求,低风险环节(如非 GMP 区域的辅助设施)可适当简化。这一原则贯穿标准始终,是所有活动的 “决策依据”。
生产系统关键要素关联:(CDEs-CAs-CPPs-CQAs )
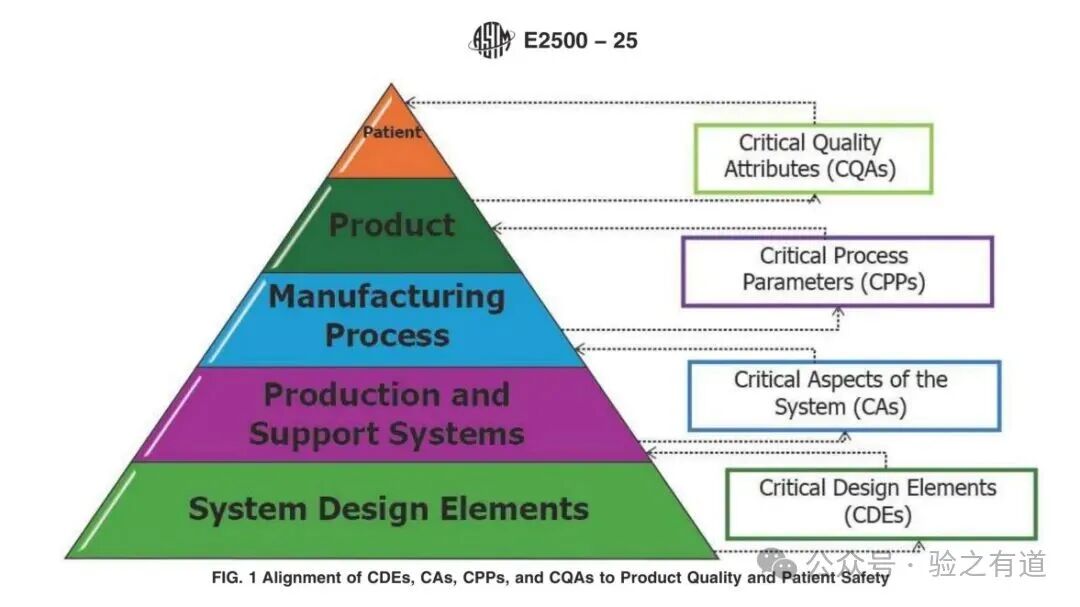
顶层目标:患者安全(Patient Safety)与产品关键质量属性(CQAs),即 “最终要保障什么”;
中间桥梁:生产工艺关键参数(CPPs),即 “通过控制哪些工艺参数实现产品质量”;
系统支撑:生产系统关键要素(CAs)与关键设计要素(CDEs),即 “系统需具备哪些特性才能稳定控制 CPPs”。
原文明确:“Critical aspects are enabled by critical design elements”(关键要素由关键设计要素支撑),而上图则直观展示了这一链条 ——CDEs 通过控制 CPPs,最终保障 CQAs,形成 “从系统设计到患者安全” 的完整风险控制路径8。例如,对于注射剂生产中的 “无菌灌装系统”,CDEs 可能包括 “灌装针头材质(防脱落)”“灌装速度控制系统(防污染)”,这些要素通过控制 “灌装精度(CPP)”,最终保障 “产品无菌性(CQA)”,进而守护患者安全。
"Quality by design concepts should be applied to ensure that critical aspects and their associated CDEs are designed into systems during the specification and design process."
(应应用设计质量理念,确保在规格制定和设计过程中,将关键要素及其相关关键设计要素融入系统设计。)
这意味着在系统设计阶段,需基于 QbD 理念完成风险评估,明确 CDEs 并写入设计文档,避免后期因 “设计缺陷” 导致验证失败。例如,在冻干机设计阶段,需提前确定 “搁板温度均匀性(CDE)”,而非在安装后才发现温度偏差影响产品稳定性。
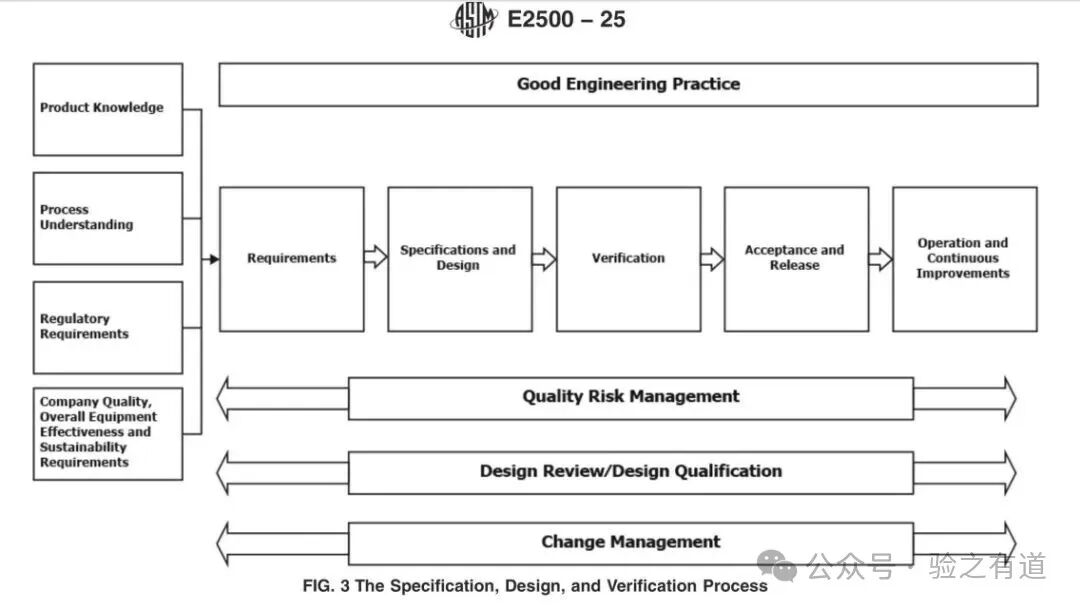
该图展示了 “规范制定 - 设计 - 验证” 的全流程,而 “良好工程实践(GEP)” 是贯穿该流程的基础。原文定义:
"Good engineering practice (GEP) should underpin and support the specification, design, and verification activities."(良好工程实践应支撑并保障规格制定、设计和验证活动。)
从图中可见,GEP 与 “产品知识、工艺理解、法规要求、质量风险管理” 共同构成流程输入,指导 “设计评审 / 设计确认(DR/DQ)”“调试(Commissioning)”“资质认定(C&Q)” 等环节,最终输出 “验收与放行(Acceptance and Release)” 结果。实操中,GEP 体现为 “标准化的工程文档(如设计图纸、调试记录)”“规范化的供应商管理(如审计)”,确保所有活动可追溯、可复现。
主题专家(SMEs)与系统所有者(System Owner)
"SMEs are defined as those individuals with specific expertise and responsibility in a particular area or field... System owner is accountable/responsible for the overall system delivery."(主题专家指在特定领域具备专业知识和责任的个人…… 系统所有者对系统的整体交付负责。)
在流程中,SMEs 需参与 “风险评估”“验证方案制定”“结果审核” 等关键环节,确保专业判断贯穿始终;系统所有者则作为 “单一责任点”,协调设计、验证、维护等全周期活动,避免责任分散。例如,在冻干机 PQ 阶段,需由 “工艺工程师(SME)” 制定冻干曲线验证方案,“系统所有者” 跟踪方案执行进度并协调解决异常。
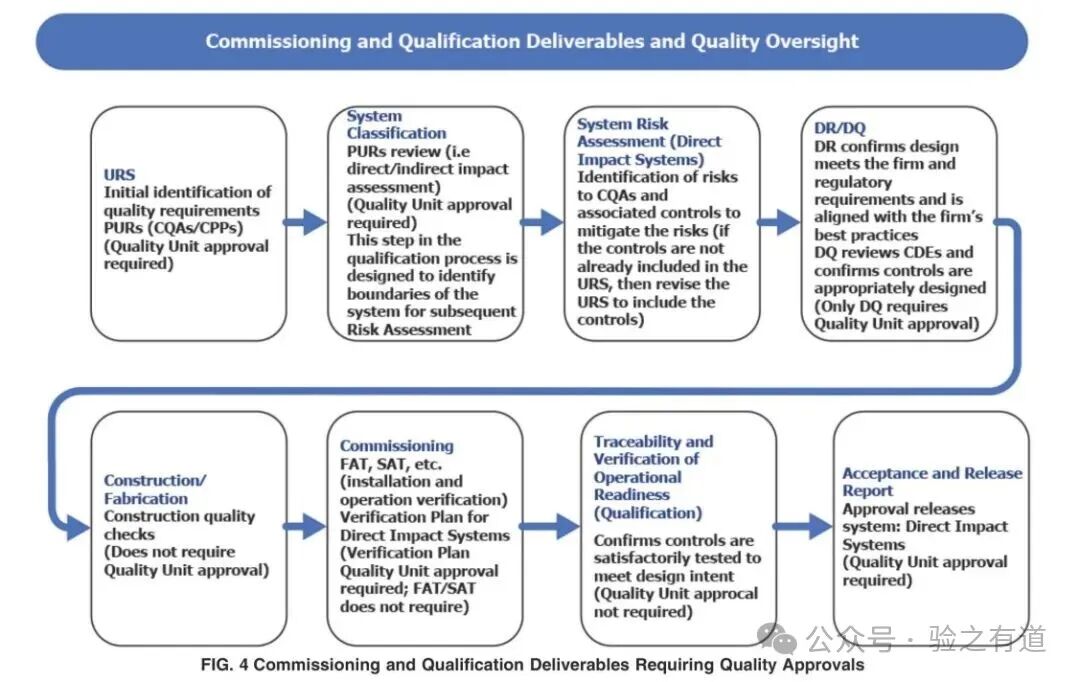
图中明确了 “哪些交付物需质量部门(Quality Unit)审批”,是持续改进的 “合规边界”。原文要求:
"As experience is gained in commercial production, opportunities for improvements should be sought based on periodic risk review and evaluation."(随着商业生产经验的积累,应基于定期风险审查和评估寻求改进机会。)
从图中可见,“设计确认(DQ)”“验证方案(Verification Plan)”“关键要素验收报告(Acceptance Report for Critical Aspects)” 需经质量部门批准,而 “调试记录(Commissioning Records)” 则无需预批准 —— 这一区分既保障了关键环节的合规性,又为持续改进预留了灵活性。例如,企业可基于生产数据,在不影响关键要素的前提下,优化设备运行参数(如调整输送速度),仅需通过 GEP 变更管理,无需重复质量审批流程。
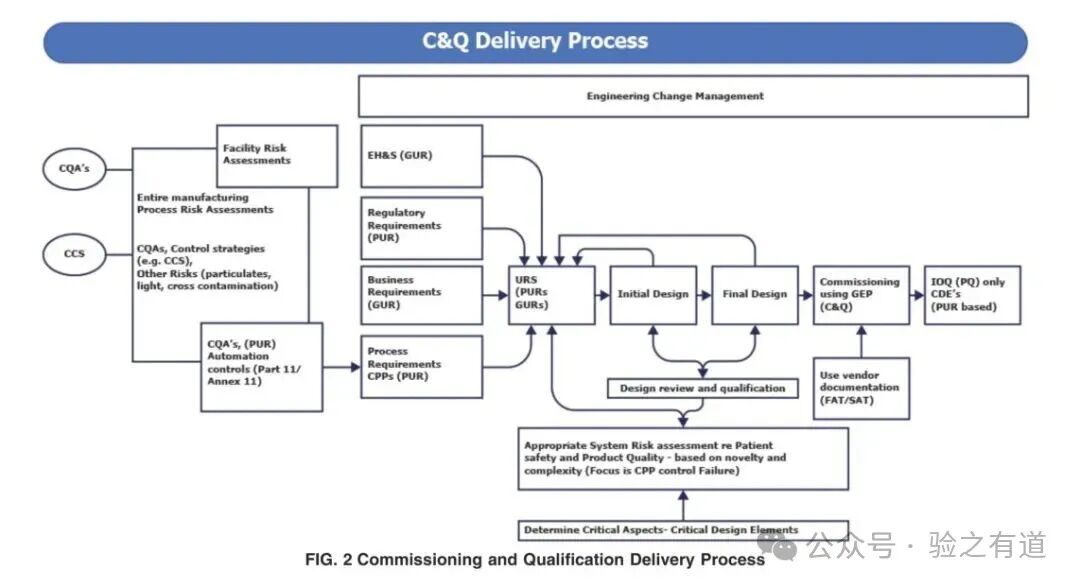
标准构建了以质量风险管理(QRM)为核心的四阶段实施流程,而上图则展示了 “从初始设计到最终验收” 的阶段划分,每一步均需形成完整文档记录。
需求定义(Requirements Definition)
"These specific requirements relative to product quality and patient safety should be based upon: Product knowledge (CQAs), Process understanding (CPPs), Regulatory requirements (QTPP), Company quality requirements (QTPP), and Known risks."
(这些与产品质量和患者安全相关的具体要求应基于:产品知识(关键质量属性)、工艺理解(关键工艺参数)、法规要求(质量目标产品概况)、公司质量要求(质量目标产品概况)及已知风险。)
从上图可见,需求定义阶段需区分 “工艺用户要求(PUR,影响产品质量)” 和 “通用用户要求(GUR,如 EHS、产能)”,前者直接关联后续 CDEs 的识别,后者通过 GEP 管理。实操要点:质量部门需早期参与,确保需求符合法规要求(如 FDA 工艺验证指南)。
规格制定与设计(Specification & Design)
核心是 “怎么设计”,关键输出为设计确认(DQ),原文要求:
"Design qualification (DQ) is a documented process that verifies that the proposed design of the facilities, systems, utilities, and equipment is suitable for the intended purpose."(设计确认是一项书面化流程,用于验证所提议的设施、系统、公用设施和设备设计适用于预定用途。)
图中显示,该阶段需完成 “初始设计(Initial Design)” 到 “最终设计(Final Design)” 的迭代,通过设计评审(DR)确认设计符合 PUR 和 GUR。实操要点:DQ 文档需经质量部门批准,重点确认 CDEs 是否满足风险控制要求(如无菌设备的 “密封完整性设计”)。
"Commissioning should include verification actions to confirm that the equipment and systems have been properly installed and are operating correctly. Qualification should include verification of critical aspects and critical design elements."(调试应包括验证活动,确认设备和系统已正确安装且运行正常。资质认定应包括对关键要素和关键设计要素的验证。)
从图上可见,调试阶段需完成 “工厂验收测试(FAT)”“现场验收测试(SAT)”,验证设备安装和基础功能;资质认定阶段则聚焦 “安装确认(IQ)”“运行确认(OQ)”“性能确认(PQ)”,仅针对 PUR 相关的关键要素。实操要点:调试可复用供应商文档(如 FAT 报告),但需先评估供应商质量体系;PQ 需证明设备在实际生产条件下稳定控制 CPPs。
验收与放行(Acceptance & Release)
"The documentation should contain a clear statement as to whether or not the manufacturing system is fit for the intended purpose,based on the review."(文档应基于审核结果,明确说明生产系统是否适用于预定用途。)
图上显示,该阶段需汇总所有验证结果,形成 “验收报告”,其中关键要素的验收需经质量部门批准。实操要点:验收文档需包含 “不合格项处理记录”(如调试中发现的 “传感器偏差” 及纠正措施),确保所有问题闭环后再放行。
风险适配性:验证的深度、文档的详略程度需与风险等级匹配,高风险环节(如直接影响 CQA 的系统)需更严格的控制措施(如增加 PQ 测试次数),低风险环节可简化流程(如复用供应商调试记录),这一原则需在图 2 的流程中明确标注。
变更管控:系统变更需对照图 1 的 “CDEs-CAs-CPPs-CQAs 链条”,判断是否影响关键要素 —— 若仅调整非关键参数(如设备外观颜色),通过 GEP 变更管理即可;若变更 CDEs(如更换灌装针头材质),则需重新评估风险并补充验证,变更文档需引用图 3 的 “设计评审流程”。
ASTM E2500-25 标准的核心价值,不仅在于明确了 “做什么”,更通过图 1、图 2、图 3、图 4 等关键图表,清晰展示了 “怎么做”“各环节如何关联”。从 “CDEs 到患者安全” 的关联链(图 1),到 “设计到验收” 的全流程(图 2),再到 “质量监督边界” 的划分(图 4),每一张图表都是合规操作的 “可视化指南”。
对于制药企业而言,遵循该标准不仅是满足国际法规要求的 “必修课”,更是提升系统可靠性、降低质量风险的 “护城河”。将标准要求与图表逻辑转化为日常 SOP,让科学风险管理贯穿设备全生命周期,才能真正实现合规与质量的双重保障 —— 毕竟,每一套合规系统的稳定运行,都是对患者用药安全的郑重承诺。
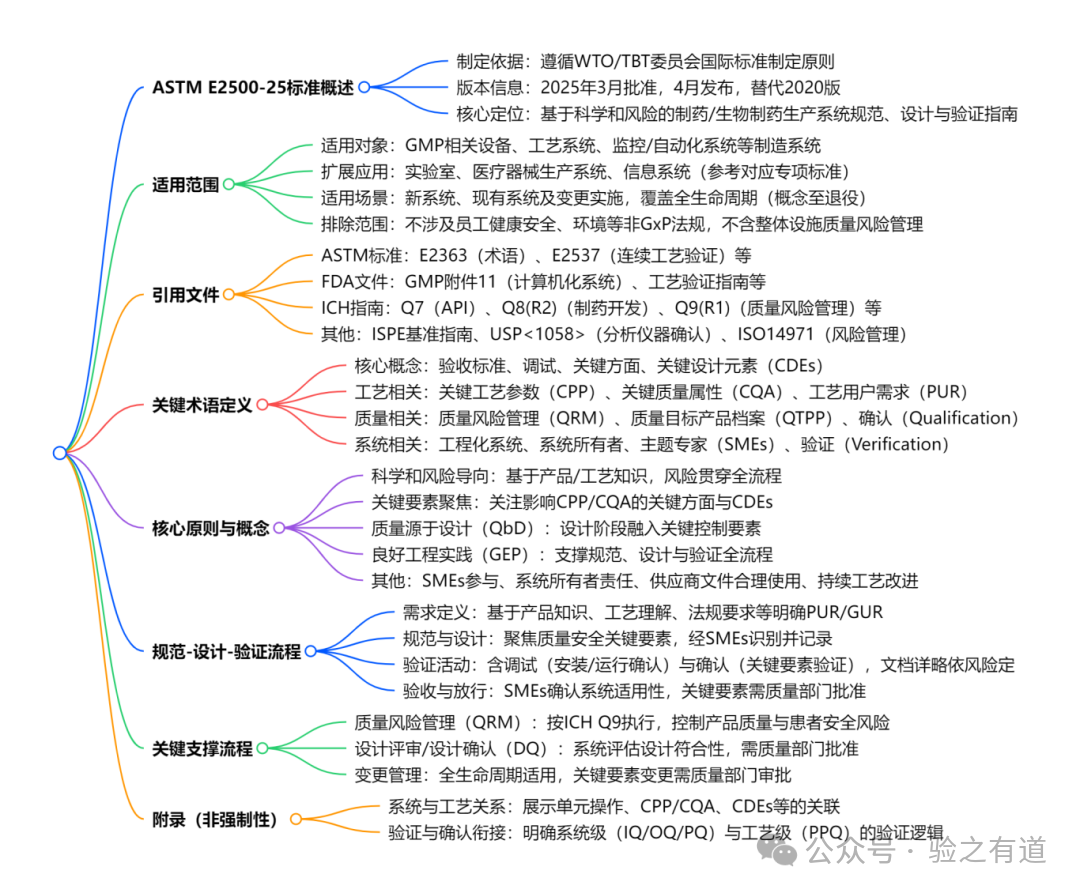
ASTM E2500-25标准脑图:


































请先 登录后发表评论 ~